
Hóa sinh của bệnh Alzheimer Biochemistry of Alzheimer’s disease
Hóa sinh của bệnh Alzheimer Biochemistry of Alzheimer’s disease
Hoàng Thị Bích Ngọc
Hội Hóa Sinh Y học Việt Nam
Tóm tắt
Bệnh Alzheimer đã được xác định là một bệnh do sự gấp nếp sai lệch của protein, do sự tích tụ của các protein Amyloid-beta trong não. Bệnh Alzheimer cũng được coi là một chứng bệnh tauopathy do sự kết hợp bất thường của protein tau, một protein liên quan đến microtubule biểu hiện trong nơ-ron , thường hoạt động để ổn định các microtubule trong bộ khung tế bào. Bộ não là cơ quan chuyển hoá một lượng lớn glucose để sản xuất năng lượng tế bào ở dạng ATP.
Sự phụ thuộc vào glucose làm cho não bị nguy hiểm nếu cung cấp glucose bị gián đoạn, hoặc khả năng chuyển hóa glucose trở nên khiếm khuyết. Sự giảm chuyển hóa glucose trong não tương quan với mật độ mảng bám và thâm hụt nhận thức ở bệnh nhân bệnh tiên triến. Những bất thường ty thể đã được tìm thấy cùng với stress oxy hoá. Stress oxy hóa liên kết không thể tách rời với một số quá trình bệnh lý trong bệnh Alzheimer, bao gồm Abeta gây ra nhiễm độc thần kinh, bệnh lý TAU, rối loạn chức năng ty thể.
Bệnh Alzheimer (Alzheimer disease –AD) là một chứng mất trí phổ biến nhất, được phát hiện lần đầu tiên năm 1906 bởi Alois Alzheimer (bác sĩ người Đức). Hiện trên thế giới, Alzheimerảnh hưởng đến khoảng 10% các cá nhân 65 tuổi, với tỷ lệ này tăng gấp đôi mỗi năm năm lên đến 80 tuổi. Năm 2010 có khoảng 35,6 triệu người trên toàn thế giới. Dự tính 20 năm sau ( 2030 là 65,7 triệu người). Riêng ở Mỹ 2012 có 5,4 triệu người là nguyên nhân thứ sáu gây tử vong. Việt Nam chiếm khoảng 5% dân số trên 60 tuổi, tỷ lệ tăng dần theo tuổi. Các phương pháp điều trị hiện tại chỉ giúp giảm một phần nhỏ triệu chứng bệnh, bệnh không thể chữa khỏi, là áp lực rất lớn về mặt xã hội, tâm lý, sức khỏe, kinh tế đối với cuộc sống của những người chăm sóc. Ở các nước phát triển, Alzheimer là một trong những bệnh tốn kém nhất cho xã hội.
Các nghiên cứu theo chiều dọc của những người bình thường tiếp tục phát triển Alzheimer cho thấy có sự chuyển đổi đột ngột về sự suy giảm triệu chứng nhận thức giữa giai đoạn tiền lâm sàng và giai đoạn sớm của Alzheimer. Sự suy giảm dần dần về mặt nhận thức trong giai đoạn tiền lâm sàng đã đạt đến một điểm uốn, dẫn tới sự mất dần khả năng nhận thức, đó là dấu hiệu của Alzheimer lâm sàng. Thông thường, sự mất khả năng nhận thức tương đối nhanh là điều làm cho các thành viên trong gia đình hoặc người chăm sóc đưa bệnh nhân khám bệnh. Bởi thời gian bệnh nhân đến để chẩn đoán đã có quá nhiều tổn thương não không thể đảo ngược với bất kỳ phương pháp điều trị để có hiệu quả
Bệnh được biểu hiện bởi sự có mặt của các mảng bám amyloid betaᵦ (A) ngoại bào và các đám rối tơ thần kinh (NFTs ) nội bào , làm suy giảm chức năng của các tế bào thần kinh và gây chết tế bào.Sự thoái hoá thần kinh được ước tính bắt đầu từ 20-30 năm trước khi bất kỳ có biểu hiện lâm sàng nào của bệnh trở nên rõ ràng. Các mảng bám và các đám rối tơ thần kinh nội bào đã được coi là các biểu hiện sớm của Alzheimer, dẫn đến sự thoái hóa thần kinh sau đó và apoptosis của nơ-ron.Tuy nhiên mật độ và vị trí của mảng bám amyloid không tương quan với các triệu chứng hoặc mức độ nghiêm trọng của Alzheimer.
Các phân tích giải phẫu học cho thấy các thay đổi bệnh lý lan rộng khắp vỏ não và các cấu trúc xung quanh, đặc biệt là vùng đồi thị .
Các nghiên cứu theo chiều dọc của Alzheimer cho thấy: Nồng độ amyloid ᵦ trong dịch não tủy dường như giảm 25 năm trước khởi phát bệnh . sự tich tụ amyloid ᵦ dần dần xuất hiện ở não khoảng 15 năm trước khi có triệu chứng. Mức độ tăng của protein TAU và TAU phosphoryl hóa trong dịch não tủy và chứng teo não xuất hiện đồng thời khoảng 15 năm trước khi có triệu chứng. Sự suy giảm ký ức theo giai đoạn xuất hiện 10 năm trước triệu chứng. Cuối cùng giảm nhận thức rõ ràng khoảng 5 năm trước triệu chứng.
Alzheimer được xác định là bệnh do sự gấp nếp sai lệch của phân tử protein tạo sự tích tụ protein amyloid ᵦ có các gấp nếp bất thường . Đây là những peptid ngắn . Chúng kết hợp thành các mảng amyloid. Sự tích tụ của các vi sợi (fibril) amyloid, được coi là dạng có độc tố làm ngăn cản cân bằng ion canxi trong tế bào, kích hoạt sự chết tế bào theo chương trình (apoptosis)
Alzheimer cũng được coi là một chứng bệnh tauopathy do sự phosphoryl hóa quá mức của protein TAU dẫn đến protein TAU không thể liên kết với protein liên kết- vi ống làm cho vi ống không ổn định. Các protein tau này không bị ràng buộc tạo ra sự tình trạng lộn xộn của các neurofibrin (đám rối sợi thần kinh – NFT). Các sợi này có ảnh hưởng nhiều đến chức năng nội bào
Sự hình thành mảng bám amyloid
Mảng bám amyloid là sự phân giải (amyloid precursor protein – APP) một glycoprotein xuyên màng, chức năng chưa rõ. APP gồm 770 a a, protein này biểu hiện trong nhiều mô nhưng đặc biệt trong các khớp thần kinh cuả các neuron. APP bị phân cắt bởi 3 enzym : α , ᵦ và ᵧ secretase . Trước hết APP bị cắt cạnh tranh bởi α và ᵦ secretase. Nhiều nghiên cứu cho thấy stress oxy hóa làm giảm hoạt tính của α-secretase , làm tăng cường biểu hiện và kích hoạt β-secretase. Con đường α-secretase tạo ra sAPPα , các sAPP α hòa tan này liên quan đến sự dẫn truyền thần kinh bình thường ở synap và C83 . Con đường β-secretase sinh ra sAPPβ và C99 . Các sản phẩm C83 và C99 này bị cắt một lần nữa nhờ ᵧ secretase (ᵧ secretase là một phức hợp đa enzyme có chứa presenilin 1,2. Đột biến presenilin được tìm thấy ở bệnh nhân Alzheimer sớm có tính chất gia đình). Riêng với C99 dưới tác dụng của ᵧ secretase sẽ tạo thành hai phân đoạn không hòa tan Amyloid ᵦ 40 (A ᵦ40) và Amyloid ᵦ 42 (A ᵦ42). A ᵦ42 có tính chất độc hơn và nhiều hơn A ᵦ40. Các monomer này trải qua một sự thay đổi đáng kể về cấu hình ở nồng độ cao để hình thành cấu trúc bậc ba của dạng tấm beta . Những mảnh này sau đó kết hợp lại thành các oligomer (là dimer hoặc trimer), chúng dường như là các chất độc thần kinh, không hòa tan xung quanh các tế bào thần kinh và dần dần thành các mảng amyloid beta ( hình 1).
Ngoài Amyloid beta, mảnh protein tiền chất vùng nội bào (fragment: amyloid precursor protein intracellular domain – AICD) cũng có tác dụng phá vỡ sự truyền tin giữa các nơ-ron trong quá trình tiến triển của bệnh Alzheimer. Nội dung này đã được Pousinha trình bày những phát hiện này tại Hội nghị Khoa học thần kinh học diễn ra từ ngày 17 đến 21/ 10 /2015 tại Chicago.-
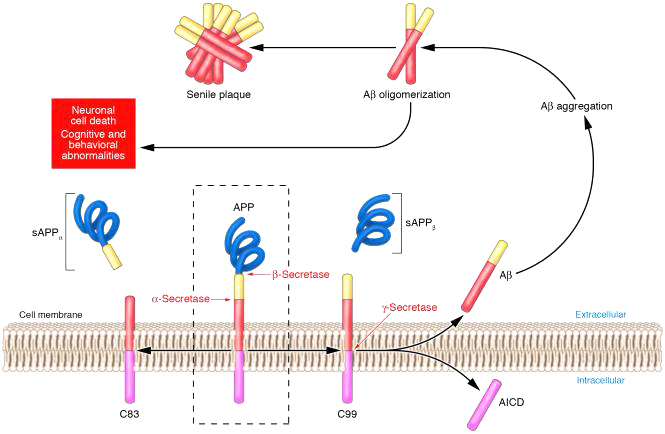
Hình 1. Sự hình thành Amyloid beta và mảng bám ngoài tế bào
Sự hình thành đám rối sợi thần kinh (neurofibrillary tangles- NFT) do protein TAU
Protein TAU là một protein liên kết vi chất được tìm thấy trong hầu hết các mô và tồn tại rất cao trong hệ thần kinh trung ương và ngoại vi. Nó là thành phần quan trọng của bộ khung tế bào thần kinh để ổn định các vi ống, vi ống rất cần thiết cho việc duy trì cấu trúc của neuron, vận chuyển của sợi trục, và sự dẻo dai của synap. Protein TAU có 6 đồng phân, phụ thuộc vào sự tồn tại của ba hoặc bốn vị trí liên kết với tubulin, được quy định bởi trạng thái phosphoryl hóa, có khoảng 79 vị trí có khả năng phosphoryl hóa trên đồng phân TAU dài nhất . Ở protein TAU bình thường có khoảng 30 vị trí phosphoryl hóa. Thông qua các đồng phân của protein TAU , sự phosphoryl hóa của nó tương tác với tubulin để ổn định sự lắp ráp vi ống. Ở giai đoạn phôi thai, protein TAU chủ yếu được tìm thấy trong trạng thái tăng phosphoryl hóa, do nhu cầu lớn về sự thay đổi trong nơ-ron thần kinh và các khớp thần kinh ở giai đoạn phát triển ban đầu của hệ thần kinh trung ương. Tuy nhiên trạng thái phosphoryl hóa vẫn thấp hơn so với bệnh nhân Alzheimer. Mặt khác, ở người trưởng thành, hệ thần kinh trung ương trưởng thành, protein TAU được dephosphoryl hoá (một quá trình ngược lại) là cần thiết cho sự ổn định của vi thể, duy trì sự cân bằng nội môi tế bào, cả ở cấp độ cấu trúc và chức năng.
Phosphoryl hóa quá mức protein TAU
Trong não bình thường TAU có sự cân bằng giữa phosphoryl hóá ( bởi các enzyme kinase) với sự O-GlcNAcyl hóa ( hình 2a) . Trong Alzheimer nhiều yếu tố : môi trường, chuyển hóa chất, di truyền gây ra thiếu glucose hoặc giảm chuyển hóa glucose dẫn đến giảm O-GlcNAcyl, quá trình phosphoryl hóa TAU sẽ tăng ( hình 2b). Trong gây mê, hạ thân nhiệt phosphatase giảm gây tăng phosphoryl hóa ( hình 3). Trong Alzheimer sự tăng quá trình phosphoryl hóa protein TAU có thể do các đột biến làm thay đổi chức năng và sự biểu hiện quá mức của các đồng phân TAU. Cũng có thể là kết quả của quá trình phosphoryl hóa. Đó là đột biến FTDB-17 tại các điểm G272V, P401L, V337M.. làm tăng lượng protein TAU bị phosphoryl hóa.
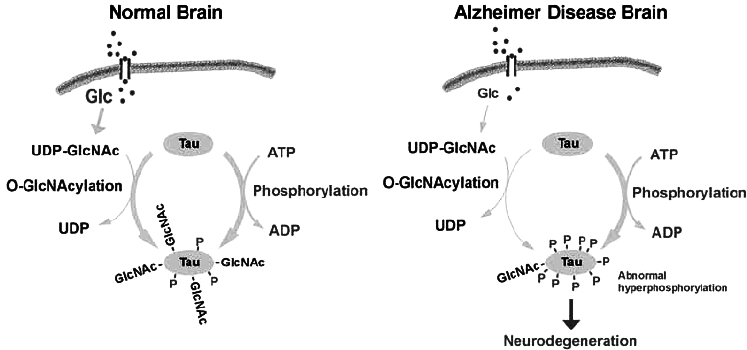
Hình 2. Sự cân bằng quá trình O-GlcNAcyl hóa và phosphoryl hóa protein tau ở người bình thường và sự mất cân băng, phosphoryl hóa quá mức protein TAU trong Alzheimer
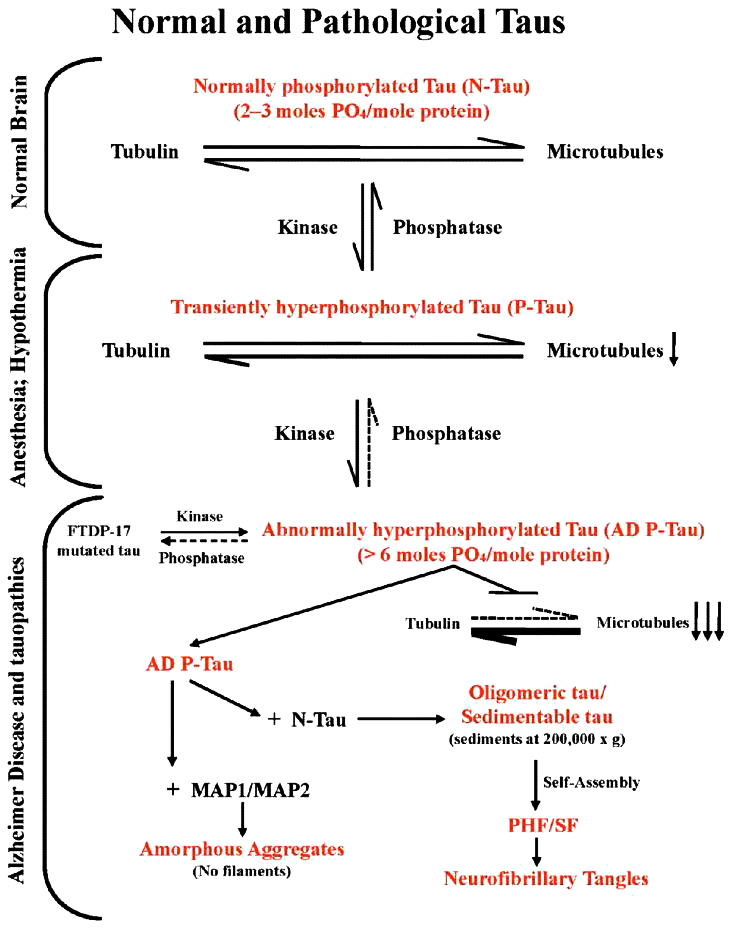
Hình 3 . Sự phosphoryl hóa TAU quá mức
Sự hình thành đám rối sợi thần kinh NFTs
Các protein TAU bị phosphoryl hóa quá mức sẽ không còn gắn kết bền vững với các tubulin . Chúng sẽ tách rời khỏi các vi ống trùng hợp thành oligomer ( khoảng 2 hoặc 3 monomer ) tạo thành kẹp đôi soắn ốc (PHFs), PHFs kết hợp với các sợi thẳng tạo thành đám rối sợi thần kinh NFTs ( hinh 4) Cơ cấu này phá hoại các chức năng của tế bào chất và cản trở vận chuyển của sợi trục, . Những thay đổi này cuối cùng dẫn đến sự sụp đổ của bộ khung tế bào, mất khả năng sống của tế bào dẫn đến sự chết của tế bào. Do đó, ức chế sự phosphoryl hoá bất thường của tau mang lại một mục tiêu điều trị đầy hứa hẹn cho Alzheimer .
Thực nghiệm trên chuột , Lithium đã được chứng minh là làm giảm sự phosphoryl hóa tau [ Điều trị bằng lithi đã được chứng minh là làm giảm mật độ rối loạn dây thần kinh trong mô hình chuyển gien ở vùng hippocampus và tủy sống. Mặc dù mật độ NFTs giảm, sự mất của bộ nhớ không được cải thiện sau khi điều trị.và Lithium cũng không có tác dụng phòng ngừa Alzheimer
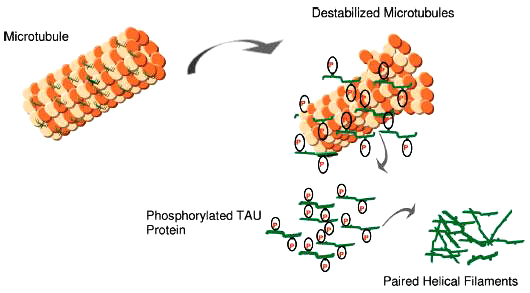
Hình 4.Trong Alzheimer, có một sự giảm khả năng ràng buộc vi ống và lắp ráp vi ống. hyperphosphoryl hóa có thể góp phần vào sự phát triển của mạng lưới sợi nhỏ, vận chuyển sợi trục không ổn định, và cuối cùng là sự hình thành đám rối sợi thần kinh (NFT)
Ngày càng trở nên rõ ràng rằng Alzheimer là một bệnh đa chức năng và đa nhân tố, có thể bao gồm các cơ chế sinh bệnh khác nhau, tuy nhiên kết quả lâm sàng tương tự. Rất nhiều yếu tố có thể coi là nguyên nhân hoặc ảnh hưởng đến Alzheimer.
Giảm chuyển hóa glucose
Nhiều nghiên cứu in vitro, in vivo và các nghiên cứu lâm sàng trên người đã cung cấp bằng chứng rằng Đái tháo đường typ 2 là một yếu tố nguy cơ chính trong bệnh lý Alzheimer. Chuyển hóa glucose bất thường là một hiện tượng sinh lý bệnh học được quan sát thấy ở Alzheimer. Trong các nghiên cứu thực nghiệm cũng cho thấy β amyloid hoạt động như một chất ức chế cạnh tranh của sự gắn kết và hoạt động của insulin ở não . Nồng độ Amyloid β trong Alzheimer có thể liên quan đến sự đề kháng insulin ở não. Với tính chất vật lý và cấu tạo phân tử , tính chất chức năng và vị trí của nơron trong các mạch thần kinh có ảnh hưởng đến số phận của chúng trong quá trình lão hóa. Các nơron chiểu phổ rộng lớn với các sợi trục dài bị tổn thương nhiều nhất trong Alzheimer. Những nơron này có đặc điểm: Nhu cầu năng lượng cao, tỷ lệ trao đổi chất cao, năng lượng phụ thuộc vào lượng glucose, có diện tích bề mặt tế bào lớn làm tăng khả năng tiếp xúc với các điều kiện môi trường độc hại. Trong nghiên cứu về sự giảm chuyển hóa glucose trong các vùng não của bệnh nhân Alzheimer, Fucuyama và cs đã nghiên cứu PET trên bệnh nhân Alzheimer, đã phát hiện sự tiêu thụ oxy, sử dụng glucose, lưu lượng máu thấp tại các vùng trán , đỉnh, thái dương của những người bị bệnh Alzheimer. Từ những kết quả này người ta cũng gợi ý rằng sự chuyển hóa glucose bất thường ở vùng đỉnh thái dương (parietotemporal region) có thể là nhân tố chính dẫn đến rối loạn chức năng tổng hợp làm ảnh hưởng đến não của bệnh nhân Alzheimer . Hiện nay kỹ thuật chụp quang phổ phát xạ 2. -[18F]fluoro-2-deoxy-D-glucose (2. -[18F]fluoro-2-deoxy-D-glucose positron emission tomography-FDG PET) là một công cụ vô giá trong chẩn đoán Alzheimer. Kỹ thuật chụp FDG PET không chỉ giúp việc chẩn đoán mà còn có ý nghĩa trong chẩn đoán sớm.
Não là nơi chỉ sử dụng năng lượng từ glucose. Sự suy giảm đáng kể trong việc sử dụng glucose ở não được thấy ở chứng mất trí của Alzheimer là kết quả của các rối loạn chuyển hóa trong sự thoái hóa glucose theo con đường đường phân (glycolysis) và sự oxy hóa pyruvate. Quá trình đường phân xảy ra trong tế bào chất, còn sự thoái hóa từ pyruvat, chu trình Krebs và sự vận chuyển chuỗi điện tử giải phóng năng lượng ATP xảy ra trong ty thể. Chuyển hóa glucose được điều chỉnh nhiều trong ty thể (hinh 5) Các số liệu phân tích từ sinh thiết não bộ của Alzheimer cho thấy sự giảm đáng kể ty thể , trong khi DNA ty thể và protein tăng lên trong tế bào chất , người ta cho rằng có thể là sự suy thoái ty thể do tự miễn. Các gen trong ty thể được thừa kế từ mẹ, vì vậy đối với những trẻ trưởng thành có mẹ bị Alzheime, kỹ thuật FDG PET giúp chẩn đoán sớm Alzheimer cho các đối tượng này.
Sự thoái hóa từ pyruvat, chu trình Krebs và sự vận chuyển chuỗi điện tử giải phóng năng lượng ATP, cân bằng calci xảy ra trong ty thể. Những bất thường trong ty thể thường được tìm thấy cùng với tổn thương oxy hóa với sự ghi nhận bởi 8 – hydroxyguanosin và nitrotyrosin chứng tỏ ty thể đã bị hư hỏng , cùng với sự giảm đáng kể hoạt tính cytocrom oxidase ở vùng não alzheimer. Sự thiếu hụt enzyme vận chuyển điện tử dẫn đến sự gia tăng sản xuất các dạng oxy hoạt động.
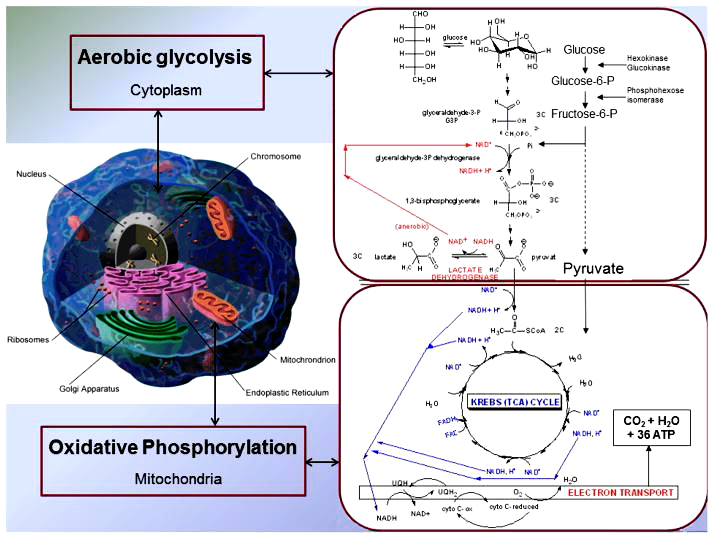
Hình 5. Chuyển hóa Glucose và chuỗi vận chuyển điện tử trong ty thể
Stress oxy hóa
Dù sự vận chuyển điện tử có hiệu quả lớn, nhưng vẫn có một số elctron bị dò dỉ sớmtạo ra các gốc tự do: dạng oxy hoạt động và các gốc tự do và các chất chống oxy hóa. Sự cân bằng này có thể bị gián đoạn trong một số tình huống bệnh lý khi các hệ thống phòng chống oxy hóa trở nên không đủ dẫn đến stress oxy hóa, dẫn đến apotosis .
Nhiều bằng chứng cho thấy sự có mặt rộng rãi trong não bộ của bệnh nhân Alzheimer như : sản phẩm hư hỏng do sự oxy hóa DNA là 8- hydrodeoxyguanin (8OHdG) và 8 hydroxyguanin (8OHG) tăng trong não Alzheimer, sản phẩm lipid peroxy hóa như malodinaldehyde (MDA), sản phẩm oxy hóa protein như 3 nitrotyrosin tăng trong não bệnh nhân Alzheimer. Trong não Alzheimer cũng thấy có sự suy giảm các enzym chống oxy hóa và Glutathion thường được tập trung ở các khớp thần kinh. Sự suy giảm này tương quan với mức độ nghiêm trọng của bệnh . Nhiều nghiên cứu đề xuất rằng tổn thương oxy hóa gây ra ở Alzheimer xảy ra trước khi xuất hiện bệnh. Stress oxy hóa có thể là một trong những thay đổi sớm nhất trong bệnh Alzheimer
Stress oxy hóa và mảng bám amyloid
Trong quá trình phân tách APP, rõ ràng hoạt động của α-secretase sẽ ngăn cản sự hình thành của Aβ42 và Aβ40 , hai chất độc hại. Stress oxy hóa làm giảm hoạt tính α-secretase trong khi đó tăng cường biểu hiện và kích hoạt ᵧ-secretase , một enzyme quan trọng trong quá trình tạo amyloid β từ APP. Sự thiếu hụt hệ thống enzyme chống oxy hóa gây tình trạng stress oxy hóa làm tăng sự lắng đọng amyloid β ở chuột đột biến biểu hiện APP. Sự thiếu hụt superoxid dismutase (Cu-Zn-SOD) trong mô hình chuột gây alzheimer làm tăng quá trình trùng hợp amyloid β và làm tăng sự mất trí nhớ về không gian của chuột. Điều này chứng tỏ sự tăng cường sản xuất amyloid β và hình thành mảng bám do stress oxy hóa .
Stress oxy hóa và phosphoryl hóa protein TAU
Stress oxy hóa được cho là xuất hiện sớm nổi bật trong quá trình sinh bệnh của alzheimer, góp phần vào sự phosphoryl hóa TAU và sự hình thành rối loạn dây thần kinh. Tuy nhiên, mối quan hệ và các cơ chế cơ bản giữa stress oxy hóa và tăng phosphoryl hóa TAU vẫn còn chưa rõ. Dữ liệu từ các thí nghiệm cho thấy rằng sự oxy hóa mãn tính làm tăng mức độ phosphoryl hóa TAU ở các sợi kép xoắn ốc (PHF-1) trong một mô hình in vitro .
Chất chống oxy hoá và phosphoryl hóa protein Tau
Một số nghiên cứu dịch tễ học đã chỉ ra mối liên quan giữa lượng chất chống oxy hoá và giảm tỷ lệ mắc chứng sa sút trí tuệ , đặc biệt là Alzheimer và suy giảm nhận thức ở người già . Trong những năm gần đây, liệu pháp chống oxy hoá đã nhận được sự quan tâm đáng kể như một phương pháp đầy hứa hẹn để làm chậm sự tiến triển của Alzheimer. Các nghiên cứu tập trung vào các chất chống oxy hoá nội sinh (ví dụ, vitamin, coenzyme Q10 và melatonin) và chất chống oxy hoá trong khẩu phần ăn như các hợp chất phenolic là flavonoid hay nonflavonoid. Sự quan tâm ngày càng tăng này đã làm tăng giả thuyết cho thấy thiệt hại do oxy hoá có thể gây ra sự suy giảm nhận thức và chức năng ở bệnh nhân alzheimer. Melatonin là một chất loại bỏ gốc tự do ngăn chặn quá trình phosphoryl hoá TAU và rối loạn vi ống trong điều kiện in vivo và in vitro . Hơn nữa, melatonin có thể là một tác nhân hữu ích trong việc phòng ngừa và điều trị alzheimer. Các chất chống oxy hoá khác, như vitamin E và C , gossypin , curcumin , beta-caroten và Ginkgo biloba cũng được báo cáo là có tính bảo vệ hiệu quả chống lại độc tính thần kinh. Demethoxycurcumin đã được chứng minh là ức chế sự phosphoryl hóa của TAU. Các thí nghiệm khác đã chỉ ra rằng thành phần hoạt tính của Ginkgo biloba, ginkgolide A ức chế phosphoryl hoá của TAU .
Từ các kết quả này cần tới những nghiên cứu thêm về hợp chất này như là một chiến lược điều trị chống oxy hóa cho Alzheimer.
Kết luận
Alzheimer là một bệnh đa chức năng và đa nhân tố, bao gồm các cơ chế sinh bệnh khác nhau, tuy nhiên kết quả lâm sàng tương tự nhau. Bệnh do sự gấp nếp sai lệch của phân tử protein tạo sự tích tụ amyloid ᵦ thành các mảng bám amylois, được coi là dạng có độc tố ngăn cản cân bằng ion canxi trong tế bào, kích hoạt apotosis. Bệnh do sự phosphoryl hóa quá mức protein TAU làm cho vi ống không ổn định. Các protein TAU không bị ràng buộc tạo thành đám rối sợi thần kinh – NFTs ảnh hưởng nhiều đến chức năng nội bào.
Não là nơi chỉ sử dụng năng lượng từ glucose thoái hóa theo con đường đường phân ái khí. Chuyển hóa glucose được điều chỉnh nhiều trong ty thể. Ty thể suy thoái cùng với tổn thương oxy hóa , tình trạng stress oxy hóa liên quan đến sự hình thành mảng bám và phosphoryl hóa TAU. Liệu pháp chống oxy hoá đã nhận được sự quan tâm đáng kể như một phương pháp đầy hứa hẹn để làm chậm sự tiến triển của Alzheimer.
SUMMARY
Alzheimer’s disease has been identified as a protein misfolding disease, or proteopathy, due to the accumulation of abnormally folded Amyloid-beta proteins in the brains of Alzheimer. Alzheimer is also considered a tauopathy due to abnormal aggregation of the tau protein, a microtubule-associated protein expressed in neurons that normally acts to stabilize microtubulesin the cell cytoskeleton.The human brain is the most organ metabolizes a large amount of glucose to produce cellular energy in the form of ATP. This dependence on glucose puts the brain at risk if the supply of glucose is interrupted, or if its ability to metabolize glucose becomes defective. diminished cerebral glucose metabolism correlates with plaque density and cognitive deficits in patients with more advanced disease.These mitochondrial abnormalities were found accompanied by oxidative stress. Oxidative stress is inextricably linked with several major pathological processes in alzheimer, including Abeta-induced neurotoxicity, tau pathology, mitochondria dysfunction.
Tài liệu tham khảo
1. C.E. Teunissen, J. de Vente, H.W.M. Steinbusch, C. De Bruijn (2002) Biochemical markers related to Alzheimer’s dementia in serum and cerebrospinal fluid; Neurobiology of Aging July–August 2002 Volume 23, Issue 4, p485-646
2. Ghulam Md Ashraf, Nigel H. Greig, Taqi Ahmad Khan, Iftekhar Hassan,Shams Tabrez,Shazi Shakil,Ishfaq Ahmed Sheikh, Syed Kashif Zaidi, Mohammad Akram Wali, Nasimudeen R. Jabir, C.K. Firoz, Aabgeena Naeem,Ibrahim M. Alhazza, Ghazi A. Damanhouri,and Mohammad Amjad Kamal (2014) Protein misfolding and aggregation in Alzheimer’s disease and Type 2 Diabetes Mellitus, CNS Neurol Disord Drug Targets. 2014; 13(7): 1280–1293.
3. Gong CX1, Liu F, Grundke-Iqbal I, Iqbal K. ( 2006) Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation, J Alzheimers Dis. 2006 Mar;9(1):1-12.
4. Lisa Mosconi( 2013) Glucose metabolism in normal aging and Alzheimer’s disease: methodological and physiological considerations for PET studies, Received: 5 March 2013 / Accepted: 25 June 2013, Italian Association of Nuclear Medicine and Molecular Imaging 2013
5. Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, Iqbal K, Gong CX (2009). Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain. 2009 Jul;132(Pt 7):1820-32. doi: 10.1093/brain/awp099. Epub 2009 May 18
6. Mushtaq G, Khan JA, Kamal MA (2014) Impaired Glucose Metabolism in Alzheimer’s Disease and Diabetes. Enz Eng 4:124. doi:10.4172/2329-6674.1000124
7. Seyedeh Maryam Alavi Naini , Nadia Soussi-Yanicostas (2015)Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies?, Oxidative Medicine and Cellular Longevity;Volume 2015 (2015), Article ID 151979, 17 pagehttp://dx.doi.org/10.1155/2015/151979
8. Yan Zhaoand Baolu Zhao(2013)Oxidative Stress and the Pathogenesis of Alzheimer’s Disease, Oxidative Medicine and Cellular Longevity Volume 2013 (2013), Article ID 316523, 10 pages
http://dx.doi.org/10.1155/2013/316523
9. Wen-Juang Huang, Xia Zhang, and Wei- Wei Chen (2016) Role of oxidative stress in Alzheimer’s disease Biomed Rep . 2016 May; 4(5): 519–522. Published online 2016 Mar 15. doi: 10.3892/br.2016.630
bài viết liên quan

Vấn đề tâm lý xã hội của tuổi già
Vấn đề tâm lý xã hội của tuổi già GS.TS. Lê Đức Hinh Chủ tịch Hội Thần kinh học Việt Nam TÓM TẮT Quá trình lão hóa, quy luật sinh học của sự phát triển, diễn ra theo một chương trình đặc hiệu cho từng cá thể, có tính quyết định tới các chức năng […]

Một số nhận xét về chứng Sa sút trí tuệ ở Việt Nam
Một số nhận xét về chứng Sa sút trí tuệ ở Việt Nam PGS.TS. Nguyễn Chương Chủ tịch Hội Thần kinh học Việt Nam, thành viên Viện Hàn lâm Thần kinh học Hoa Kỳ Khái niệm chung Là tập chứng của Thần kinh lão khoa, và cũng là phân mục của Thần kinh thoái hoá. […]

Giảng dạy và đào tạo thần kinh học tại trung tâm viện trường Bạch Mai
Giảng dạy và đào tạo thần kinh học tại trung tâm viện trường Bạch Mai PGS.TS. Nguyễn Chương Nguyên cán bộ giảng dạy Bộ môn Tinh Thần kinh từ 1960 TÓM TẮT Trước năm 1954, không có Khoa Thần kinh ở trong bệnh viện, cũng không có bộ môn Thần kinh ở Đại […]