Tin nổi bật




Hội Thần kinh học Việt Nam
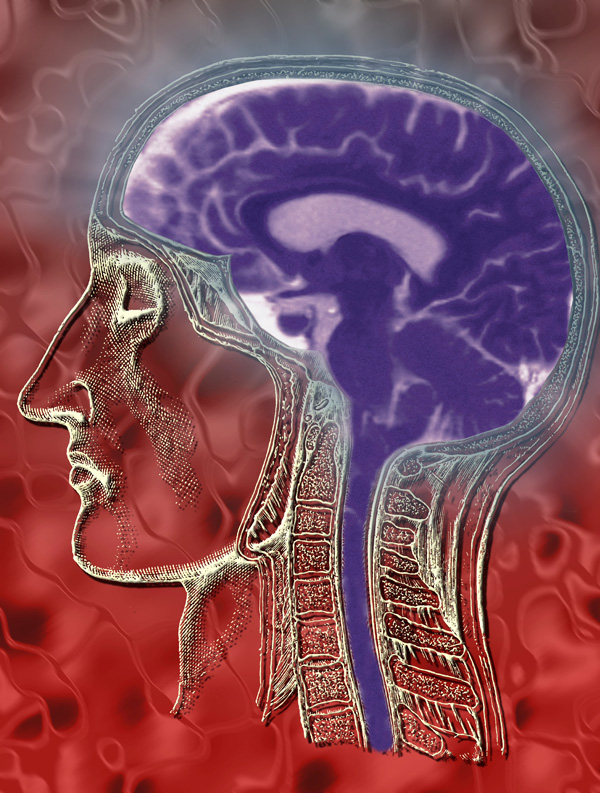
Lịch sử của Thần kinh học Việt Nam bắt đầu từ sau khi miền Bắc nước ta được hoàn toàn giải phóng tháng 10 năm 1954.
Theo Quyết định số 998/QĐ-TTg ngày 24 tháng 11 năm 1997 của Thủ tướng Chính phủ nước Cộng hòa Xã hội Chủ nghĩa Việt Nam đã giải thể Hội Thần kinh, Tâm thần và Phẫu thuật Thần kinh Việt Nam và cho phép thành lập Hội Thần kinh học Việt Nam.
Thực hiện Quyết định trên, Đại hội Đại biểu toàn quốc lần thứ nhất đã diễn ra vào ngày 20 tháng 5 năm 1998 tại Hà Nội




Tạp chí thần kinh học
Tạp chí Thần kinh học Việt Nam thuộc Hội Thần kinh học Việt Nam là diễn đàn trao đổi thông tin khoa học và hoạt động của chuyên ngành Thần kinh cả nước. Tạp chí sẽ xuất bản 4 số/năm, đăng tải các công trình nghiên cứu khoa học, bài viết, bài dịch... tạo điều kiện trao đổi học thuật, kinh nghiệm giữa hội viên và những người quan tâm tới lĩnh vực Thần kinh học.
-
Tạp chí số 41
-
Tạp chí số 42
-
Tạp chí số 43
-
Tạp chí số 44
[email protected]
Liên hệ Ban biên tập

-
Năm thành lập tạp chí
00
+ -
Số lượng bài đăng trên tạp chí
00
+ -
Số tạp chí
00
+ -
Số lượng bài đăng trên tạp chí
00
+
Nghiên cứu khoa học
Các tài liệu, bài học bổ ích về khoa Thần Kinh Học

Nghiên cứu khoa học
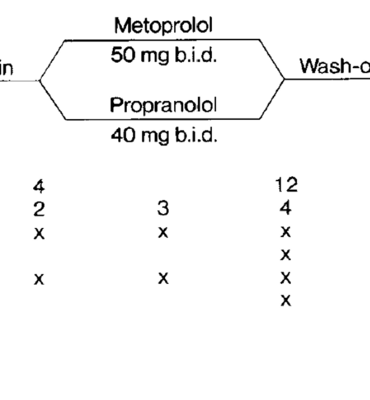
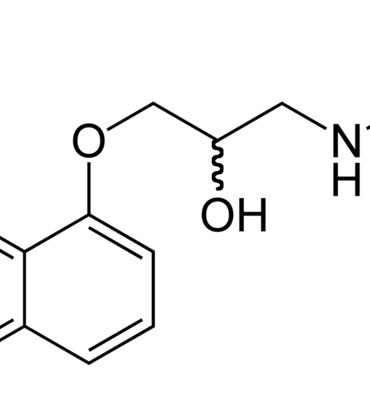
Tổng quan về các thuốc chẹn beta trong dự phòng migraine
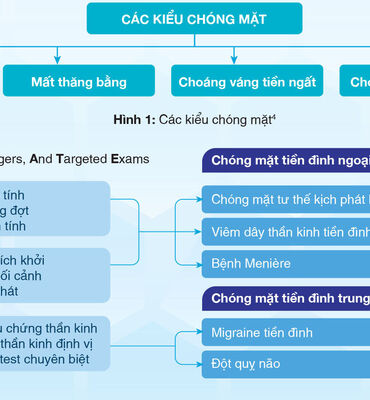
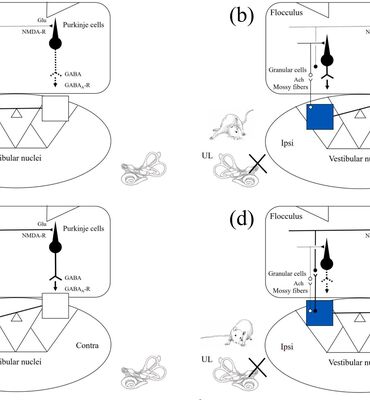
Nghiên cứu khoa học









