
Bệnh loạn dưỡng não chất trắng thượng thận : qua 3 trường hợp
Bệnh loạn dưỡng não chất trắng thượng thận : qua 3 trường hợp
Ninh Thị Ứng, Dương thùy Nga, Trần văn Học.
Tóm tắt
Bệnh loạn dưỡng não chất trắng thượng thận thường gặp và được xác định rõ ràng do sơ cứng não, tỷ lệ mắc 1/10 000 nam giới. Bệnh di truyền rối loạn tiểu thể (peroxisom) thường gặp nhất. Là bệnh thoái hóa hệ thần kinh đi kèm với suy chức năng vỏ thượng thận. Do đột biến gene trên nhiễm sắc thể X q28.
Mục tiêu: Nghiên cứu đặc điểm lâm sàng và hình ảnh tổn thương não trên MRI. Phương pháp nghiên cứu: mô tả ca bệnh.
Kết quả: BN Phạm khởi bệnh từ 5 tuổi, BN Quang từ 13 tuổi, em trai BN trung khởi bệnh từ 7 tuổi. 1 BN bắt đầu là sa sút trí tuệ; 2 BN bắt đầu là nhìn kém. Khi đã có triệu chứng lâm sàng thì MRI não đã có biến đổi. Bệnh diễn biếntrong vßng 3 n¨m, teo gai thÞ, liÖt cøng, d¸ng ®i lo¹ng cho¹ng, mÊt thÞ lùc, mÊt thÝnh gi¸c. Suy thîng thËn biÓu hiÖn b»ng s¹m da ở một BN. Một bệnh nhân được 8 lần ghép tế bào gốc máu cuống rốn.
Kết luận: Bệnh loạn dưỡng não chất trắng thượng thận chỉ gặp ở trẻ trai. Bệnh gây mất thị lực, thính giác, liệt cứng, sa sút trí tuệ. Tổn thương trên MRI não hình T2 tăng tín hiệu quanh sừng chẩm của não thất bên sau lan tỏa khắp não. Xét nghiệm huyết thanh có tăng acid béo chuỗi dài.
Đặt vấn đề
Loạn dưỡng chất trắng là một nhóm bệnh rối loạn di truyền thoái hoá thần kinh có liên quan đến tính toàn vẹn của myelin trong não và dây thần kinh ngoại vi. Hầu hết các rối loạn này gặp ở một trong ba nhóm bệnh: bệnh do Lysosomal ( bệnh tiêu thể liên quan đến chất trắng), bệnh do peroxisomal (rối loạn tiểu thể), và bệnh do mitochondrial (rối loạn chức năng ti thể biểu hiện ở não). Mỗi nhóm có đặc điểm lâm sàng, sinh hóa, bệnh học và chẩn đoán hình ảnh khác nhau.
1. Bệnh loạn dưỡng não chất trắng thượng thận (X linked adrenoleukodystrophy, X-ALD). Một ca bệnh được mô tả vào năm 1913 bởi Schilder như là “viêm não lan tỏa”; năm 1923 Siemerling và Creutzfeldt mô tả bệnh còn kết hợp teo thượng thận. 50 năm sau bệnh được phát hiện là có khiếm khuyết trong tiêu thể peroxisom beta oxy hóa có đặc điểm thừa tổng hợp acid béo chuỗi dài trong tế bào.
Bệnh X-ALD thường gặp và được xác định rõ ràng do sơ cứng não, tỷ lệ mắc 1/10000 nam giới. Là bệnh di truyền rối loạn tiểu thể (peroxisom) thường gặp nhất.Là bệnh thoái hóa hệ thần kinh đi kèm với suy chức năng vỏ thượng thận. Do đột biến gene trên nhiễm sắc thể X q28. Đã phân lập được hơn 200 đột biến.
Còn ít tác giả đề cập về bệnh này cho nên đề tài sẽ đề cập đến đặc điểm lâm sàng và hình ảnh cộng hưởng từ cũng như một số xét nghiêm cận lâm sàng qua 3 bệnh nhân.
Phương pháp nghiên cứu
Mô tả ca bệnh
Chẩn đoán dựa trên lâm sàng, hình ảnh cộng hưởng từ não, phân tích huyết thanh có tăng acid béo chuỗi dài carbon C22-C30.
Chẩn đoán phân biệt với các bệnh có loạn dưỡng não chất trắng trên cộng hưởng từ.
Kết quả nghiên cứu
Mô tả từng ca bệnh.
1.Trình bày bệnh nhân 1: A.B.Phạm sinh năm 2002. Tiền sử sản khoa bình thường. Từ nhỏ phát triển bình thường. Từ 5 tuổi, tháng 11/ 2007 trẻ xuất hiện mắt mờ, đã được chẩn đoán là bệnh X-ALD. Từ tháng 3/2008 thị lực rất kém, chỉ nhìn những vật to, đi khó dần. tăng phản xạ gân gối cả 2 chân, 2 tay yếu dần. Từ năm 2009 trẻ không đi được, không nhìn thấy, không nghe thấy, không nói được, có một số cơn co giật toàn thân.
MRI biến đổi đặc hiệu của X-ALD. MRI tháng 2/2008 thấy có sự khác thường của tín hiệu T2 siêu mạnh chất trắng quanh não thất bên, dưới vỏ của thùy đỉnh, thái dương, chẩm, vùng bao trong đều bị tổn thương, tiểu não chưa bị tổn thương. Như vậy có sự loạn dưỡng toàn phần não, loạn dưỡng cục bộ khá rõ nét ở thùy đỉnh. Bệnh tiến triển nặng vì điểm Loes 16. Xét nghiệm huyết thanh tăng acid béo chuỗi dài Cacbon 22-26. Phân tích gene phân tử không phát hiện được đột biến. Synacthen test cứ 3 tháng /1 lần điều trị Hydrocortisone 4mg/kg khi có nguy cơ suy thượng thận.
Các xét nghiệm công thức máu, sinh hóa, chức năng gan, thận, và xét nghiệm miễn dịch trong giới hạn bình thường.
Điều trị: Dinh dưỡng ít chất béo và dùng dầu Lorenzo 25-30 ml/ ngày, Flumicil, các vitamin A,D,E,K và acid béo chính, truyền gamma globulin 1-2 lần / tháng, Depakine chống co giật.
Từ 25/5/2009 đến 2 8/9 / 2009 bệnh nhân được ghép 8 lần tế bào gốc máu cuống rốn tại Trung Quốc. Quá trình làm lần I: Lấy dịch não tủy 3 ml (tốc độ 20 giọt /phút mang đi xét nghiệm), rất từ từ ỉntrathecal tiêm thuốc Dexamethasone 2mg + 2 ml tế bào gốc máu cuống rốn, tiếp tục truyền tĩnh mạch 20ml tế bào gốc máu cuống rốn. Theo dõi, để BN nằm ngửa, không cho ăn uống trong 6 tiếng. Do chức năng nuốt của họng khó khăn, thường xuyên bị viêm phổi, nên từ 2/7/2009 thông qua nội soi dạ dầy, tạo ống thông với dạ dầy, ăn qua ống thông với dạ dầy.
Bệnh nhân được theo dõi tiếp tục tai Bệnh viện Nhi trung ương từ tháng 1 năm 2010, các triệu chứng lâm sàng như đã mô tả ở trên, chưa có cải thiện sau ghép tế bào gốc.Các xét nghiệm coctísol 8 giờ sáng bình thường, tủy đồ bình thường, chức năng gan, thận bình thường. Tiếp tục điều trị triệu chứng, uống Depakine 0,5 – 1 viên / ngày, các vitamin A, D, E, chăm sóc tốt, chống bội nhiễm.
2. Mô tả bệnh nhân thứ 2. BN N.N.Quang sinh 1993. con thứ nhất.Tiền sử sản khoa bình thường, học hết lớp 7. Bệnh bắt đầu từ năm 2006 (13 tuổi) sa sút trí tuệ dần, sau 6 tháng mắt nhìn mờ, chân đi yếu từ năm 2007, năm 2008 không nói, không nhìn thấy. Biến đổi MRI não: thoái hóa myelin chất trắng của não. Hiện tại liệt cứng hai chân, nằm một chỗ. BN được điều trị ở BV Việt Pháp xác định chẩn đoán X-ALD.
3. Mô tả BN thứ 3. N.N. Trung 7 tuổi (sinh 2003). (Em ruột của BN Quang), vào BV Nhi Trung ương ngày 29 tháng 7 năm 2010, 7 ngày trước vào BV, sốt nhẹ, nôn, co giật, nhìn mờ, đọc khó khăn, chậm dần về tinh thần. Anh Trai mắc bệnh cách đây 5 năm.
Thăm khám: tiền sử sản khoa BT, từ nhỏ đến 7 tuổi phát triển bình thường. Khám: da trẻ hơi sạm, mắt nhìn mờ, phản xạ gân gối tăng cả hai chân, phản xạ Babinsky +, cơ lực yếu 2 chân, trương lực tăng, dây thần kinh sọ não không liệt. Đi bắt đầu khó khăn vì trẻ nhìn kém và liệt cứng hai chân, BN nói ít đi và thính lực BT.
Các xét nghiệm đã làm: Công thức máu trong giới hạn BT;
Protit 80,7 g/l, albumin 44,3 g/l, cholesterol 4,5 mmol/l
Magie 0,85 mmol/l, sắt 21,3 micromol/l .phóspho 1,21mmol/l, alb 0,52g/l, globulin alpha1 0,05 g/l, alpha2: 0,11g/l, beta 0,14 g/l, gamma 0,18 g/l, a/g 1,08, Triglycerit 5,1 mmol/l, ure 5,1mmol/l, creatinin 55, 1 mmol/l, đường 5,0mmol/l, SGOT 34,7 u/l, SGPT 24,6 u/l, LDH 251,2 u/l, lactic 1,9mmol/l, Chì huyết thanh 6,9 mcg/dl ( BT < 40).
Soi đáy mắt: Võng mạc mạch máu BT, gai thị hồng bờ rõ.
ACTH: 15,7 mmol/l ( BT < 10 mmol/l). cortisol 8 giờ 22,3 mmol/l giảm (BT 150mmol/l)
DNT: protein 1,22g/l, đường 3,3mmol/l , Nacl 126 mmol/l, Pandy ++, tế bào 0 BC.
Công thức máu: BC 8.400 u/l, tt 64,9%, lym. 27,8%, mono 5,4%, Hb 12,5 g/dl, TC 421.000.
Cấy DNT: PCR : CMV -, EBV (-), EV (-), HSV (-).
Điên cơ : NCV tay : 56,5 m/s , tibial 53,6 , peronal 59,7 m/s.
Siêu âm bụng BT, gan, thượng thân BT.
Mẫu máu , nước tiểu đã được gửi đi Nhật để xét nghiệm acid béo chuỗi dài chưa có kết quả.
Hình ảnh MRI: Hình T2 tăng tín hiệu mạnh ở vùng chẩm, quanh sừng chẩm của não thất bên tổn thương giai đoạn đầu của X-ALD rất đặc hiệu.
Điều trị: Không thể ghép tủy vì anh BN cũng mắc bệnh. Vì có dấu hiệu của suy thượng thận nên chỉ định Huajo (hydrocortizon ) 10mg/ 1,5 viên/ ngày ( 24kg). Vitamin A,D,E,K. Tư vấn dinh dưỡng ít chất béo, dùng dầu Lorenzo 25-30 mg/ngày, ăn chất bột.
– Tóm lại: 1 BN khởi bệnh từ 5 tuổi, 1 BN khởi bệnh từ 7 tuổi, 1 BN từ 13 tuổi: 1 BN bắt đầu là sa sút trí tuệ; 2 BN bắt đầu là nhìn kém. Khi đã có triệu chứng lâm sàng thì MRI não đã có biến đổi. Bệnh diễn biến trong vòng 3 năm, teo gai thị, không nhìn, không nghe được, liệt cứng, dáng đi loạng choạng tiến đến không đi được, mất thị lực, mất thính giác. Suy thượng thận biểu hiện bằng sạm da ở một BN. 1 BN xét nghiệm huyết thanh tăng acid béo chuỗi dài.
Bàn luận
Bệnh loạn dưỡng não chất trắng não gây nên do Peroxysom tham gia vào quá trình beta oxy hóa các acid béo có chuỗi rất dài, tổng hợp các plasmalogen, acid mật, cholesterol, oxy hóa các D amin, thải trừ H2O2 bằng các enzyme dị hóa. Bệnh của Peroxysom được chia thành 3 nhóm:
Nhóm 1: Hội chứng gan não thận của Zellweger, loạn dưỡng não chất trắng thượng thận sơ sinh, bệnh giả Réfsum trẻ nhỏ.
Nhóm 2: Bệnh biểu hiện loạn sản sụn từng điểm ở gốc chi, thiếu hụt các đa enzyme nhưng không tăng các acid béo chuỗi rất dài.
Nhóm 3: bệnh thiếu hụt acyl CoA oxydase, 3 oxy acyl CoA thiolase dẫn đến không phá hủy acid béo chuỗi dài đó là bệnh loạn dưỡng não chất trắng thượng thận liên quan đến nhiễm sắc thể X (X-ALD) và bệnh Refsum người lớn.
2. Bệnh do lysosom có tổn thương não chất trắng gồm: loạn dưỡng não chất trắng dị sắc, loạn dưỡng não chất trắng tế bào hình cầu, bệnh Nieman – Pick, bệnh Fabry, bệnh gangliosidosis GM1, GM2; mucopolysacharidosis, lipofuschin, mucolipidosis.
3.Bệnh do ty thể có sơ hóa chất trắng não: bệnh viêm hoại tử não bán cấp ( Leigh syndrome), bệnh não như tai biến, bệnh cơ, tăng acid lactic (MELAS); hội chứng động kinh giật cơ với sợi cơ đỏ nham nhở ( (MERRF syndrome), hội chứng Keans – sayre, rối loạn acid amin và acid hữu cơ, bệnh Canavan ( thoái hóa xốp hệ thần kinh).
Nhóm rối loạn não chất trắng không không rõ căn nguyên chuyển hóa như bệnh Pelizaéus – Merzbacher, bệnh Alexander, loạn dưỡng cơ bẩm sinh ( Fukuyama).
X -ALD chỉ gặp ở trẻ trai, (3 BN trên đều là nam giới) phụ nữ mang đột biến gene. Không thấy sự biến đổi song hành giữa đột biến gene và hình ảnh lâm sàng ( Powers và Moser 1998, Dubois – Dalcq 1999). Đặc điểm bệnh là tổn thương chọn lọc ở tuyến thượng thận và tổ chức não và tủy. X-ALD có 2 thể: thể trẻ em bắt đầu từ 5-10 tuổi sa sút trí tuệ, thay đổi tính tình, học kém, bệnh thoái lùi về vận động và giác quan trong vòng 3 năm, teo gai thị, liệt cứng, dáng đi loạng choạng, mất thị lực, mất thính giác, suy thượng thận biểu hiện bằng sạm da.
Thể người lớn bắt đầu từ 20-30 tuổi, (adrenomyeloneuropathy, AMN) thiểu năng thượng thận,tổn thương thần kinh tủy, tổn thương não chất trắng.
Ở trẻ em có phản ứng viêm ở chất trắng não đây là viêm tự miễn cho nên xét nghiệm dịch não tủy albumin tăng, vậy nên BN Trung khi thấy
albumin trong dịch não tủy tăng , BN được chẩn đoán là viêm não.
Nghiên cứu trên vi thể bệnh X-ALD phát hiện mất myelin trục thần kinh và tế bào ít đuôi gai, tăng hoạt hóa tế bào hình sao,muộn hơn hình thành các hốc não và tiêu tế bào thần kinh. Có hiện tượng viêm quanh mạch máu và tích tụ mỡ, có tế bào lympho, đại thực bào TNF alpha và interleukin 1.
Chẩn đoán phân biệt:
1. với loạn dưỡng não chất trắng dị sắc (metachromattic leukodỉstrophy, MLD). Thuộc nhóm bệnh do lysosom. Tỷ lệ mắc 1/40 000 trẻ sinh ra.
Tỷ lệ tử vong tuỳ thuộc từng thể, trẻ càng nhỏ bệnh càng tiến triển nhanh, ở người lớn bệnh tiến triển từ từ, âm ỉ.
Không có sự khác biệt giữa giới nam và nữ; chủng tộc. (X-ALD chỉ có nam mắc bệnh).
Lâm sàng:
Ở trẻ trước 4 tuổi: rối loạn bước đi, dừng phát triển vận động, trí nhớ mất dần, giảm sự tập trung, suy sụp dần, teo gai thị, hạn chế phản xạ sâu. Có thể bị động kinh, tử vong sau 5 năm khởi bệnh.
Trẻ từ 4-6 tuổi: rối loạn bước đi, run, vụng về, mất khả năng vận động tinh tế, suy sụp về trí tuệ, thay đổi hành vi, có thể có động kinh. Chết sau 10-15 năm phát hiện bệnh. đôi khi bị viêm tuyến mật, túi mật, viêm tuyến tuỵ, có thông báo về u bụng, chảy máu đường tiêu hoá.
Trẻ trên 6 tuổi và người lớn: Giảm khả năng học tập ở trường học, giảm trí nhớ dần, thay đổi hành vi, rối loạn tâm thần, động kinh, mất chức năng vận động tinh vi. Có tính bốc đồng, teo gai thị. ở người lớn đôi khi có vận động múa vờn.
Hình ảnh cộng hưởng từ: tổn thương não trắng và teo não.
Bệnh sinh. MLD có đặc điểm không có khả năng chuyển hoá sulfate glycolipid đặc biệt là galactosyl 3- sulfate. Trong bào quan lysosom thiếu men hoạt hoá sulfat sulfatase (arylsulfatase A). Dưới kính hiển vi ở các tổ chức có các hạt biến sắc. Thoỏi húa myelin ở trung tâm và ngoại vi, tế bào ít đuôi gai cũng mất myelin ở hệ thần kinh trung ương và ngoại vi. Sự tích tụ sulfatide dẫn đến mất chức năng nhận thức và vận động.
Điều trị: ghép tế bào gốc máu cuống rốn.
- Bệnh Canavan trẻ em (thoái hoá xốp của trục thần kinh). Bệnh do rối loạn ty thể.
- Di truyền ẩn NST thường. Đột biến khác nhau của gen mã hoá enzym
- Bắt đầu 3-6 tháng, giảm trương lực cơ dẫn đến liệt co cứng, teo thị giác, mù, múa vờn, sau xuất hiện động kinh, đầu to, có y văn công bố là không có đầu to
- Thể sơ sinh: giảm trương lực cơ, yếu, bú kém, chết trong vài tuần
- Thể trẻ nhỏ: bất đầu dưới 5 tuổi, loạn choạng tiểu não, run, liệt co cứng, thoái hoá võng mạc, sắc tố võng mạc, teo thị giác.
– Cộng hưởng từ não, trên T2 giảm tỉ trọng não lan toả chất trắng (N.acetyl aspartat tập trung trong não)
– Điều trị: Ghép tủy máu cuống rốn.
– Tiên lượng chết lúc 3-4 tuổi
– Thiếu men; Aspartoacylase
– Test chẩn đoán: thiếu men trong tế bào tổ chức liên kết. thử nước tiểu với N acetyl aspartate. Cộng hưởng từ não, trên T2 giảm tỉ trọng não lan toả chất trắng (N.acetyl aspartat tập trung trong não) (hình 2)
- Bệnh Alexander
- – Di truyền lẻ tẻ nam nhiều hơn nữ
– Bắt đầu từ lúc đẻ đến 2 tuổi, liệt cứng, co giật, đầu to có thể kết hợp với não ứng thuỷ
– Thể xuất hiện muộn hơn: bắt đầu từ 7-14 tuổi có triệu chứng hành tuỷ, liệt cứng, loạn choạng tiểu não,trí tuệ bình thường
– Thể người lớn: lâm sàng như xơ cứng rải rác nhiều nơi.
– MRI: thoái hóa lan tỏa não chất trắng.
– Điều trị: ghộp tủy mỏu cuống rốn.
– Tiên lượng: chết dưới 5 tuổi
– Khuyết tật sinh hoá chưa rõ
– Chẩn đoán bệnh sinh có sợi Rosenthal
Bệnh loạn dưỡng não chất trắng có tế bào hình cầu (Krabbe)
– Di truyền ẩn NST thường
– Bắt đầu từ 3-8 tháng, tăng kích thích, sốt, tăng trương lực cơ, bệnh nhân phát triển liệt cứng, teo thị giác, mù điếc, co giật, có thể tổn thương dây thần kinh, hiếm có bớt đỏ.
– Thể tuổi dậy thì và tuổi người lớn, loạn tinh thần tiến triển chậm, mù, liệt, cứng.
– MRI : Hình T2 : tăng tín hiệu ở thùy đỉnh, tiếp theo là xung quanh não thất bên, quanh thể trai, thùy chẩm. ( X-ALD tăng tín hiệu ở thùy chẩm trước).
– Điều trị: Ghép tế bào gốc máu cuống rốn.
– Tiên lượng: chết sau 1 năm khởi phát bệnh
– Thiếu men Galactocerebroside-beta-galactosidase
– Test chẩn đoán: thử men trong tế bào bạch cầu
Ảnh MRI của bệnh X-ALD.
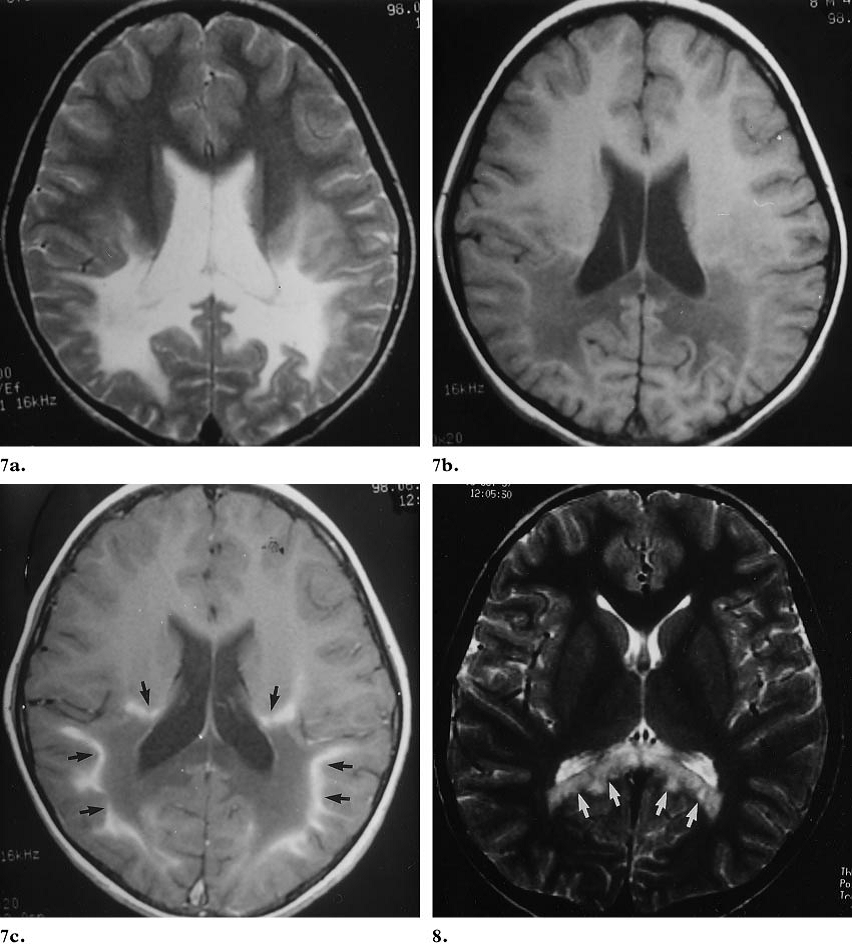
Giai đoạn đầu, huỷ hoại đối xứng chất trăng 2 bên, xuất hiện ở vùng quanh não thất bên của sừng chẩm. sau đó tiếp tục tiến triển lên phía trên đến khi toàn bộ chất trắng bị phá huỷ. Vùng vỏ não chất trắng bị tổn thương ở giai đoạn muộn. Vùng trung gian thể hiện hoạt động viêm và phá vỡ hàng rào máu não. T2 vùng này thể hiện hình ảnh đồng tín hiệu hoặc hơi giảm nhẹ tín hiệu và bắt thuốc sau khi tiêm thuốc cản quang. Vùng ngoại vi giới hạn phạm vi hủy hoại myelin, thể hiện tăng tín hiệu T2. Hình ảnh điển hình tăng tín hiệu bất thường đối xứng 2 bên ở bó tháp dưới.
Hình 2. MRI của bệnh Canavan thoái hóa sốp chất trắng não lan tỏa cả hai bán cầu, teo vỏ não.( chẩn đoán phân biệt với X-ALD)
Kết luận
Bệnh loạn dưỡng não chất trắng thượng thận chỉ gặp ở trẻ trai. Bệnh gây mất thị lực, thính giác, liệt cứng , sa sút trí tuệ.
Tổn thương trên MRI não hình T2 bắt đầu tăng tín hiệu quanh sừng chẩm của não thất bên sau lan tỏa khắp não ( thoái hóa myelin).
Xét nghiệm huyết thanh tăng acid béo chuỗi dài.
Summary
X liked adrenoleukodistrophy: Report 3 cases.
Introduction: Adrenoleukodystrophy is the most common and clearly defined form of sudanophilie cerebral sclerosis, with an incidence of
1/10 000 male subjects. It is also the most common inherited peroxisomal disorder. The gene whose mutation cause the disease has been mapped to chromosome Xq28.
Objective: To study clinical and MRI of X-ALD.
Method: We report 3 cases.
Results: First 17 years boy now, this was from 13 years old presented by psychomotor regression 6 months prior and then visual impairment, one year later lost of vision and hearing. His brother onset of X-ALD from 7 years with visual impairment. The third patient onset from 5 years with visual impairment, gradual disturbance in gait, spastic contractures of the lower extremities. One patient have cutaneous melanosis – adrenal insufficiency. One patient have chemical analysis demonstrates the accumulation of very long chain fatty acid with carbon chain lengths of 22 to 26 but could’t determine the nature of the mutation. T2 Weighted MRI are first seen in the periventricular posterior parietal and occipital regions and extend anterorly with progression of the diseases. One patient had been the haematopoietic stem cell transplantation.
Conclution: X-ALD onset in boy, with visual, intellectual impairment, spasticity. On MRI demyelinating lesions in the periventricular posterior and abnormally high levels of plasma very long chain fatty acid.
Tài liệu tham khảo.
1.Gerard Ponsot, Michel Arthus, Nicole Pinsard. Neurology pediatrics, Maladies metaboliques et heredo- degeneratives, Medecine Sciences Flammarion, 1990 : 518-560.
2.Kliegman behzman Jenson Stanton. Nelson textbook of pediatrics, The nervous system, Degenerative brain diseases, 18th edition, Saunders Elsevier 2007: 1333-1374.
3.Jean Aicardi, Cheryl Hemingway, Hermione lyall , Heredodegenerative disorders, 3rd. edition, Wiley Blackwell 2009: 327 – 381.
4.John H. Menkes. Child Neurology, Diseases with degeneration primarily affecting white matter, sixth edition, Lippincott Williams & Wilkins, 2000: 211- 240.
bài viết liên quan

Nghiên cứu tác dụng hỗ trợ điều trị động kinh của chế phẩm Egaruta
Nghiên cứu tác dụng hỗ trợ điều trị động kinh của chế phẩm Egaruta GS.TS. Nguyễn Văn Chương*, ThS.BS. Đỗ Đức Thuần*, BSCKI. Đào Hùng Vương* *Bộ môn – Khoa Thần kinh – Bệnh viện Quân y 103 – Học viện Quân y TÓM TẮT Mục tiêu: nghiên cứu tác dụng hỗ trợ điều trị động […]

Nghiên cứu tác dụng của chế phẩm Tuệ Đức An Giấc Nữ ở bệnh nhân nữ rối loạn giấc ngủ
Nghiên cứu tác dụng của chế phẩm tuệ đức an giấc nữ ở bệnh nhân nữ rối loạn giấc ngủ GS.TS. Nguyễn Văn Chương, ThS. BS CKI. Đỗ Đức Thuần Bộ môn Nội Thần kinh –Bệnh viện Quân y 103 – Học viện Quân y Tóm tắt Nghiên cứu thử nghiệm lâm sàng ngẫu nhiên […]
Đau sau đột quỵ não từ lâm sàng, hình ảnh học đến cập nhật điều trị
Đau sau đột quỵ não từ lâm sàng, hình ảnh học đến cập nhật điều trị TS.BS. Đinh Hữu Hùng Bộ môn Thần kinh, Khoa Y Dược, Trường ĐH Tây Nguyên Tầm quan trọng của đột quỵ não Gánh nặng về kinh tế-xã hội và sức khỏe của người dân do các không lây nhiễm, […]